

Services
NGS
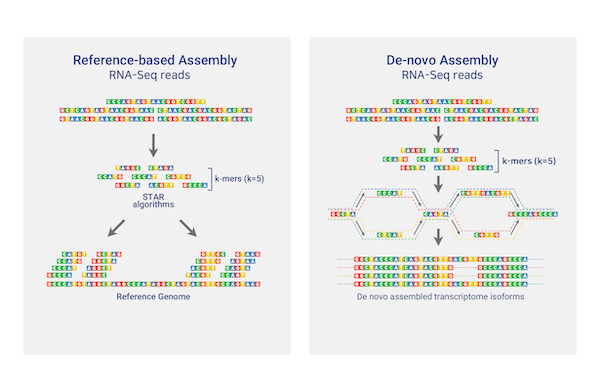
Total/mRNA Sequencing
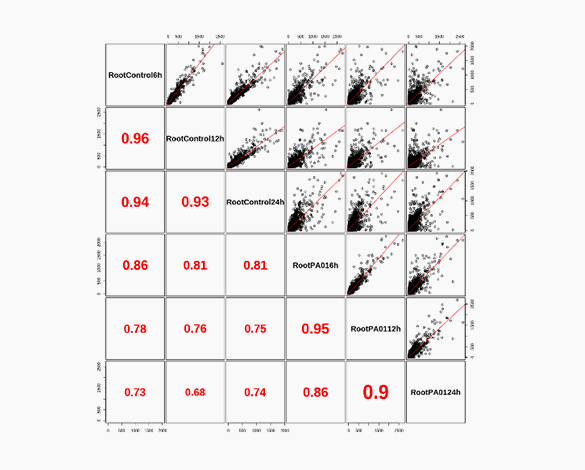
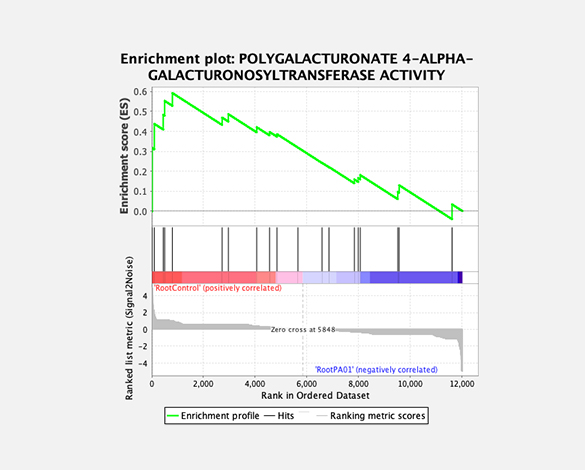
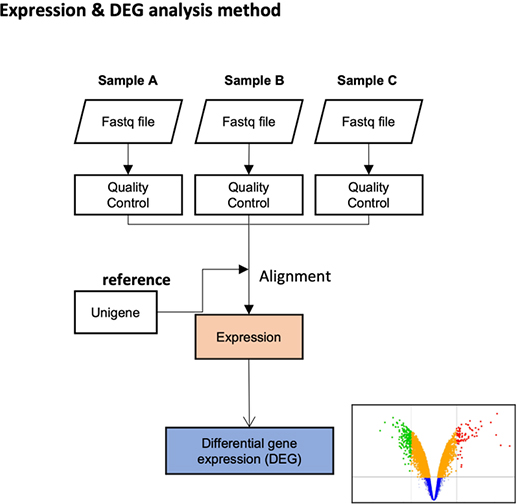
Theragen Bio provides differentially Expressed Gene (DEG) analysis between normal and control groups from various samples such as human, organoids, plants, and xenocrafts andbased on this analysis, we provide the necessary basis for gene function research. Gene set enrichment analysis (GSEA) is possible to identify genes related to biological characteristics that have statistical significance under certain conditions (e.g., normal cells vs cancer cells). In addition, we also provide information on previously unidentified transcripts, alternative splicing, and gene fusion
Sample/Laboratory Information
| Sample Requirement | 0.1 ~ 1µg Total RNA 0.1 ~ 4µg Total RNA, RIN ≥ 8 |
|---|---|
| Library Kit | Illumina, TaKaRa, Qiagen |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | 20M (million reads) |
Bioinformatics
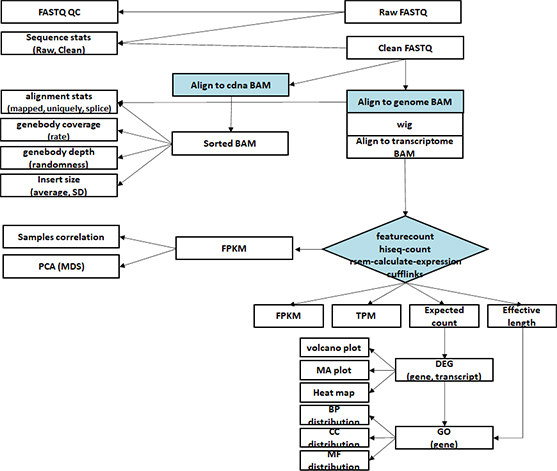
- Workflow
-
-
Standard Analysis
Functional annotation
Alternative splicing analysis
RNAseq De novo assembly
Differential expression genes (DEGs)
GO analysis
Heatmap
-
Advanced Analysis
Gene set enrichment analysis (GSEA)
Gene fusion analysis
No content
No content
No content
Features & Benefits
- Obtain extensive data at the gene level that can capture all biological changes
- Possible to obtain known or unknown information at the gene level
- Quantitative measurement at the gene level is possible even without reference genome information.
Reference
[1] Nakayama, M. et al., (2020). Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nature communications, 11(1), 2333.
[2] Hong, E. et al., (2020). Inhibition of TGF-β signalling in combination with nal-IRI plus 5-Fluorouracil/Leucovorin suppresses invasion and prolongs survival in pancreatic tumour mouse models. Scientific reports, 10(1), 2935.
[3] Cha, S. et al., (2021). Differential activation mechanisms of two isoforms of Gcr1 transcription factor generated from spliced and un-spliced transcripts in Saccharomyces cerevisiae. Nucleic acids research, 49(2), 745–759.
[4] Yun, S. M. et al., (2013). PPP1R1B-STARD3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of PI3K/AKT signaling. Oncogene, 33(46), 5341–5347. https://doi.org/10.1038/onc.2013.472
[5] Yoon, K. et al., (2013). Comprehensive genome- and transcriptome-wide analyses of mutations associated with microsatellite instability in Korean gastric cancers. Genome Research, 23(7), 1109–1117. https://doi.org/10.1101/gr.145706.112
[6] Kim, K. et al., (2018). Pathway profiles based on gene-set enrichment analysis in the honey bee Apis mellifera under brood rearing-suppressed conditions. Genomics, 110(1), 43–49. https://doi.org/10.1016/j.ygeno.2017.08.004
[7] Sakai, E. et al., (2017). Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Research, 78(5), 1334–1346. https://doi.org/10.1158/0008-5472.can-17-3303
[8] Park, Y. et al., (2020). Destablilization of TRAF6 by DRAK1 Suppresses Tumor Growth and Metastasis in Cervical Cancer Cells. Cancer Research, 80(12), 2537–2549. https://doi.org/10.1158/0008-5472.can-19-3428
[9] Lee, J. et al., (2021). Brassinosteroid‐BZR1/2‐WAT1 module determines the high level of auxin signalling in vascular cambium during wood formation. New Phytologist, 230(4), 1503–1516. https://doi.org/10.1111/nph.17265
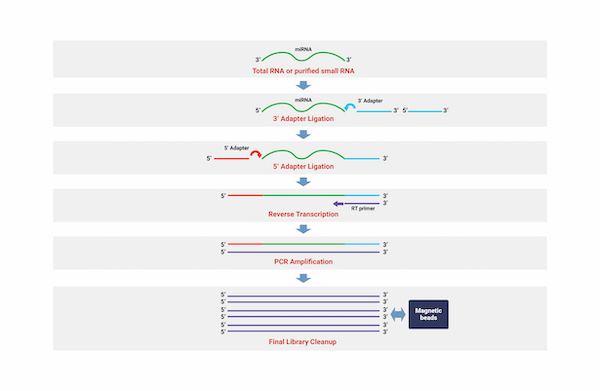
Small RNA Sequencing
Through the technology of identifying small non-coding RNAs (ncRNAs) that regulate various biological processes such as cell growth, tissue differentiation, cell death, and viral infection, we can analyze thousands of small ncRNA sequences such as micro RNAs (miRNAs) on a broad scale. We provide researchers with information on small RNA units that are differentially expressed within samples, including miRNAs that are not yet known. We explore miRNA seed area mutations and measure their expression levels using genetic information for the respective species. Additionally, we provide information on the genes targeted by miRNAs by integrating various database information and information on the genes targeted by each miRNA.
Sample/Laboratory Information
| Sample Requirement | Total RNA > 1µg (minimum 100 µg), 20 ng/µl, RIN > 6 |
|---|---|
| Library Kit | PerkinElmer |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | ≥ 20 million read counts per sample |
Bioinformatics
- Workflow
-
-
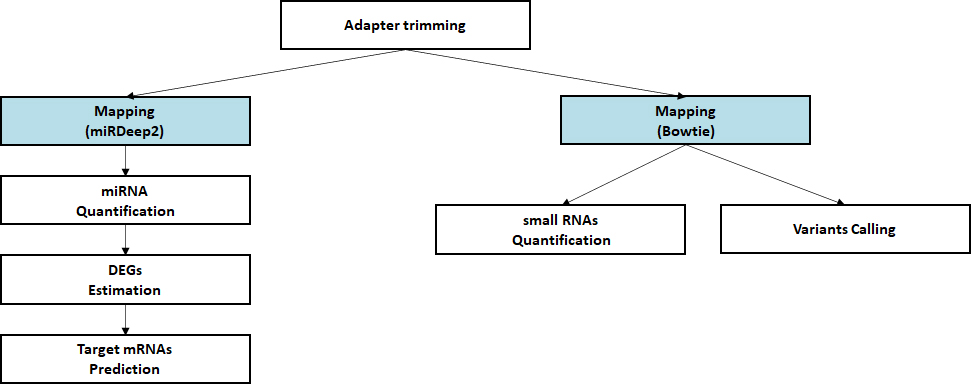
Standard Analysis
Alignment against reference genome
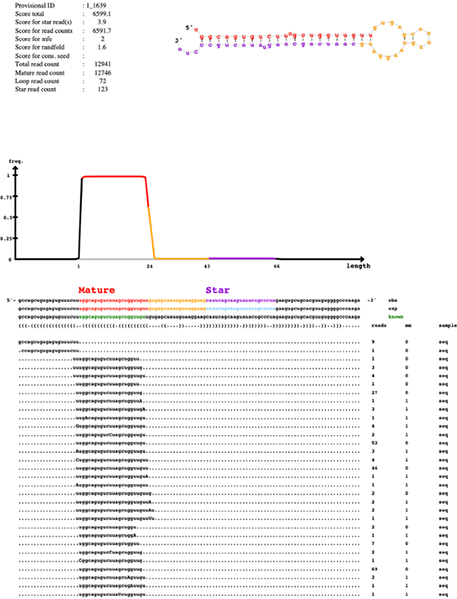
Quantification of small RNA
Discovery of novel/known miRNAs
Estimation of differential expressed miRNAs
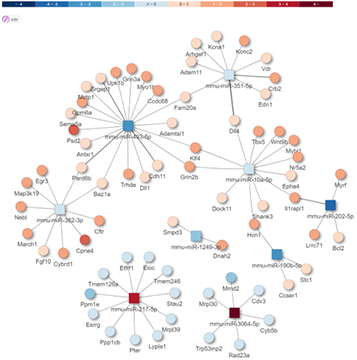
Prediction of target mRNA
-
Advanced Analysis
miRNA-mRNA Integrated analysis
No content
No content
No content
No content
No content
Reference
[1] Lee, J. Y., Ryu, D. S., Kim, W. J., & Kim, S. J. (2016). Aberrantly expressed microRNAs in the context of bladder tumorigenesis. Investigative and clinical urology, 57 Suppl 1(Suppl 1), S52–S59.
[2] Kim, M. C. et al., (2014). Identification and characterization of microRNAs in normal equine tissues by Next Generation Sequencing. PloS one, 9(4), e93662.
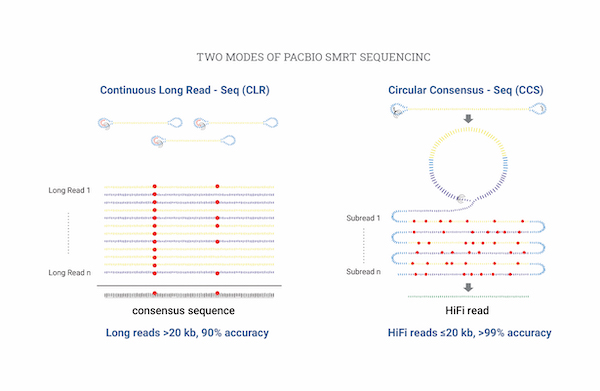
Isoform Sequencing (de novo)
Isoform sequencing allows for sequencing and analysis of full-length isoforms while preventing errors that may arise from short-read sequencing. By using PacBio-based long-read sequencing, clustering, and annotation, unigenes can be created. Additionally, Illumina-based short-read sequencing can be used to calculate RNA-seq expression for unigenes.
Application
- Alternative splicing
- Alternative polyadenylation (APA)
- Fusion transcripts
- Long non-coding RNAs (lncRNAs)
- Isoform phasing
- Genome annotation
Sample/Laboratory Information
| Sample Requirement | RNA > 3µg (min 1µg),100ng/µl, RIN > 8 |
|---|---|
| Library Kit | SMRT |
| Sequencing Platform | PacBio Sequel Ⅱ, PacBio Revio |
| Recommended Sequencing Depth | ≥ 15Gb per sample ≥ 100 million read pairs per sample *Depth may vary depending on species |
Bioinformatics
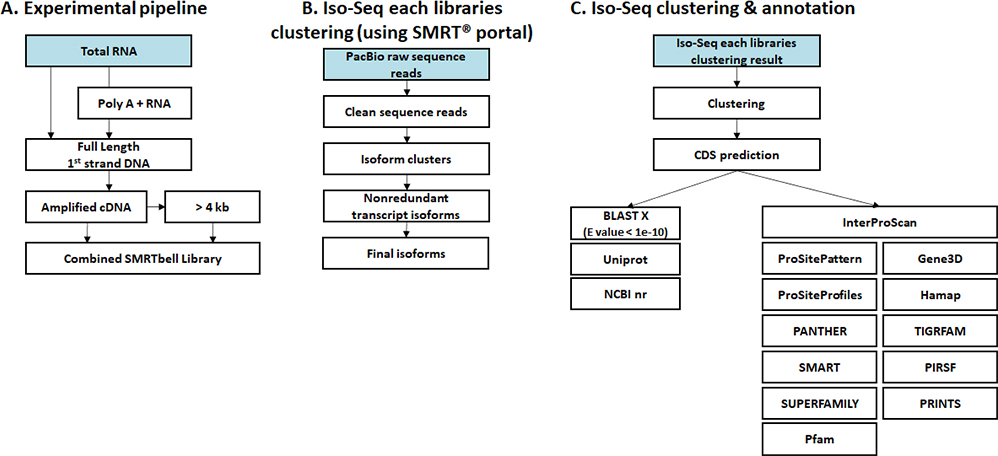
- Workflow
-
Standard Analysis
Clustering & Annotation
Read mapping
Expression
Differential expression genes (DEGs)
Reference
[1] Jo, I. H. et al., (2017). Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome. Genes, 8(9), 228.
[2] Hong, C. P.et al., (2022). Long-read transcriptome sequencing provides insight into lignan biosynthesis during fruit development in Schisandra chinensis. BMC genomics, 23(1), 17.

TCR Sequencing
TCR Sequencing is an immune profiling technology that analyzes the genetic sequences of T-cell receptors (TCR) to study T-cell diversity and characteristics. This allows for the examination of the TCR repertoire, which reflects how effectively the immune system can respond to various antigens. Additionally, TCR sequencing is utilized to investigate changes in T-cell diversity and characteristics under pathological conditions, playing a crucial role in tracking immune responses in diseases such as immune disorders, infections, and cancer. Theragen Bio's TCR Sequencing solution features both proprietary TCR Assay and an Analysis Platform, ensuring high efficiency not only with PBMC samples but also with FFPE samples.
Application
- Immune Checkpoint Inhibitor
- Cell Therapy
- Cancer Vaccine
- Senescence
- Infectious Diseases
- Hematopoietic Stem Cell Transplantation (HSCT)
Sample/Laboratory Information
| Sample Requirement | Samples extracted from PBMC, T cells, and Fresh Frozen Tissue : Total RNA ≥0.5ug, ≥35ng/ul, RINA ≥5 Samples extracted from FFPE : Total RNA ≥0.5ug, ≥35ng/ul, RINA ≥1.5, DV200 ≥70% |
|---|---|
| Library Kit | TB-TCR beta Assay |
| Sequencing Platform | Illumina NovaSeq6000, Illumina NovaSeq X Plus |
| Recommended Sequencing Depth | ≥ 3Gb per sample |
Bioinformatics
- Workflow
-

-
Standard Analysis
Clonal Diversity
Rarefaction Curve
Clonotype Proportion (Nucleotide / Amino Acid)
CDR3 Length Distribution
Gene Arrangement
Annotation of the Clonotypes

Reference
[1] IgBLAST : Ye, Jian, et al. “IgBLAST: an immunoglobulin variable domain sequence analysis tool.” Nucleic acids research 41.W1 (2013): W34-W40.
[2] VDJtools : Shugay, Mikhail, et al. “VDJtools: unifying post-analysis of T cell receptor repertoires.” PLoS computational biology 11.11 (2015): e1004503.
[3] Immunarch : Team, ImmunoMind. “immunarch: an R package for painless bioinformatics analysis of T-cell and B-cell immune repertoires.” Zenodo 10 (2019): 5281.
- Theragen Bio
- Business Number : 816-86-01640
- CEO : Jin Yeob, Go / Soon Myung, Paik
- B-4F, 145, Gwanggyo-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do, Republic of Korea
- Tel : +82+1522-2382
- Fax : +82-31-288-1293
COPYRIGHT(C) THERAGEN BIO CO.,LTD. ALL RIGHTS RESERVED.
