

Services
NGS
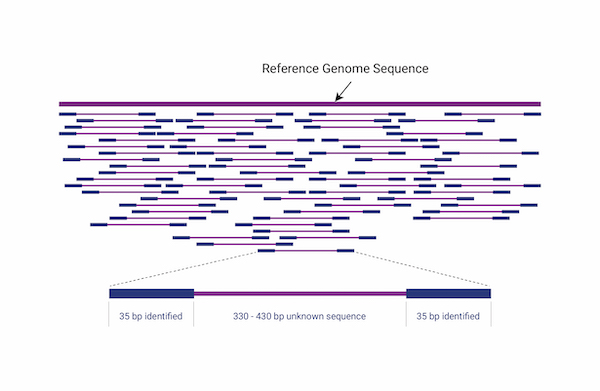
Whole Genome Resequencing
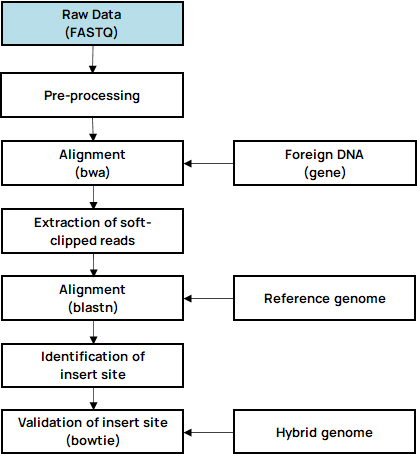
Whole genome sequencing is a comprehensive analysis of the entire genome, providing analysis of genetic variation across the entire genome, including germline, somatic mutations, copy number variants(CNV), and structural variants(SV). For non-model species, whole genome resequencinganalysis(WGRS) is performed through customized analysis, including analysis of reference size and annotation information after consultation.Integration site analysis, which identifies the location of vectors (or integrated genes) inserted into host genome DNA using bioinformatics technology, is an important aspect of genetic recombination research as vectors can affect the expression of target genes or oncogenes depending on their type and integration method.
To analyze next-generation sequencing (NGS) data quickly and accurately, Theragen Bio has introduced the DRAGEN Bio-IT Platform. This platform provides a bioinformatics pipeline optimized for mapping, alignment, duplication marking, haplotype variant calling, and customized analysis for germline, somatic, RNA, single-cell RNA, methylation, joint genotype analysis, and DRAGEN-GATK. Germline-based WGS sample analysis takes about 40 minutes to 1 hour per sample and can analyze many samples simultaneously. For somatic analysis, 300 samples can be analyzed over a period of 3 days, with each sample taking approximately 1 hour to analyze. Theragen Bio was the first in Korea to successfully operate Illumina ICA and has analyzed over 5,000 WGRS samples to date, achieving successful results in AWS cloud environment.
Application
- Population genetics
- Genetic rare diseases
- Cancer research
Sample/Laboratory Information
| Sample Requirement | gDNA > 500ng (min 100 ng), 5 ng/µl, DIN > 6 |
|---|---|
| Library Kit | Illumina, PacBio |
| Sequencing Platform | Illumina NovaSeq6000, PacBio Sequel Ⅱ |
| Recommended Sequencing Depth | For rare disease >30X |
Bioinformatics
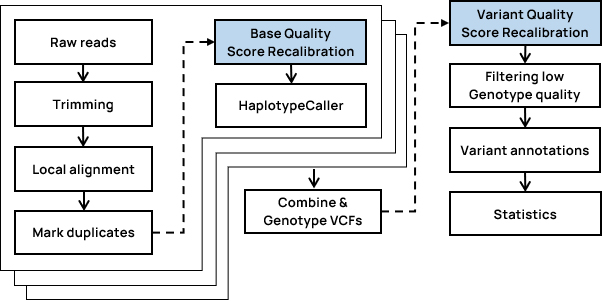
- WGRS Workflow
- Insertsite Workflow
-
Standard Analysis
Read mapping
Variants calling
Variant annotation
No content
-
Advanced Analysis
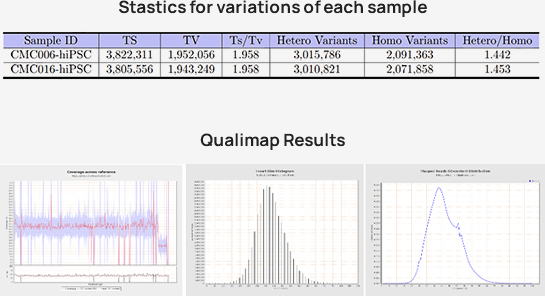
Somatic variants calling
Copy number variants(CNV)
Structure variants(SV)
Multisample variants calling
Integration site analysis
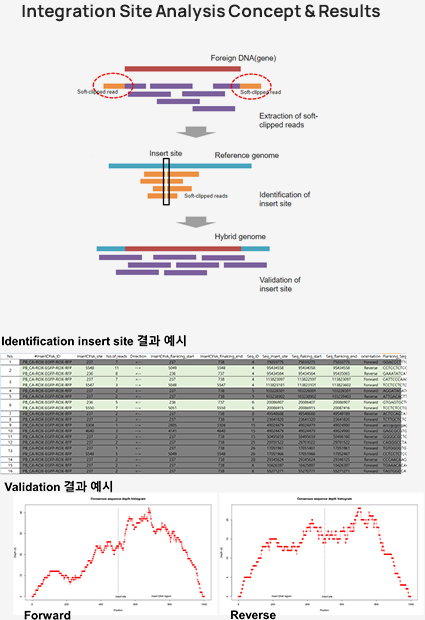
Features & Benefits
- Experience in WGRS analysis of various model and non-model species.
- Experience in large-scale analysis such as the Korean Chip Project.
Reference
[1] Ahn, S. M. et al., (2009). The first Korean genome sequence and analysis: full genome sequencing for a socio-ethnic group. Genome research, 19(9)
[2] Gallego Llorente, M. et al., (2015). Ancient Ethiopian genome reveals extensive Eurasian admixture throughout the African continent. Science (New York, N.Y.), 350(6262), 820–822.
[3] Kim, M. Y. et al., (2010). Whole-genome sequencing and intensive analysis of the undomesticated soybean (Glycine soja Sieb. and Zucc.) genome. Proceedings of the National Academy of Sciences, 107(51), 22032–22037. https://doi.org/10.1073/pnas.1009526107
[4] Yoon, K. et al., (2013). Comprehensive genome- and transcriptome-wide analyses of mutations associated with microsatellite instability in Korean gastric cancers. Genome Research, 23(7), 1109–1117. https://doi.org/10.1101/gr.145706.112
[5] Xu, X. et al., (2013). The Genetic Basis of White Tigers. Current Biology, 23(11), 1031–1035. https://doi.org/10.1016/j.cub.2013.04.054
[6] Li, E. et al., (2020). Neural stem cells derived from the developing forebrain of YAC128 mice exhibit pathological features of Huntington’s disease. Cell Proliferation, 53(10). https://doi.org/10.1111/cpr.12893
Whole Genome De novo Sequencing
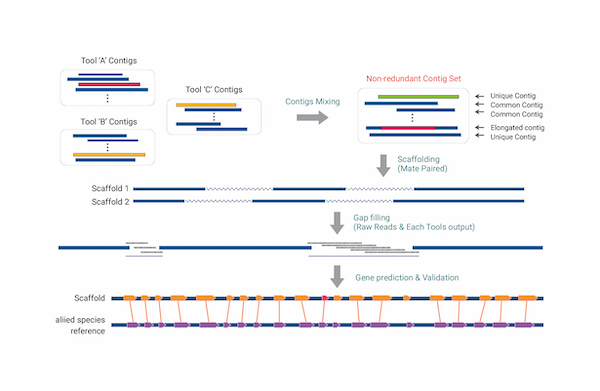
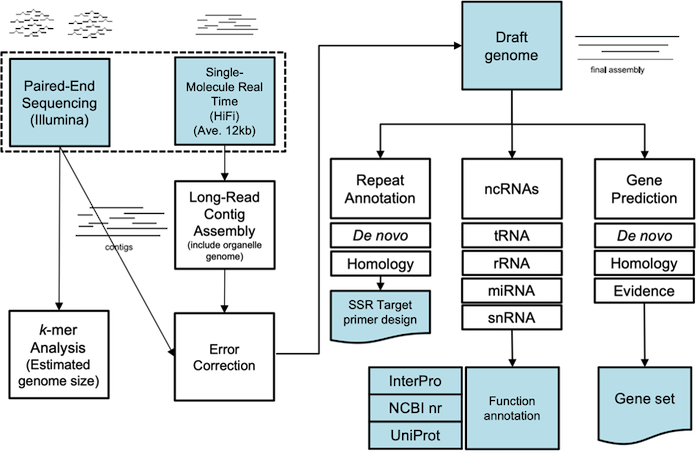
Whole-genome de novo sequencing using PacBio long-read sequencing technology is mainly focused on animal, plant, and microbial genomes to secure genetic resources. Generally, the results of research on food resources are reported.Recently, standard genome assembly analysis has been conducted to obtain genome information on useful genetic resources, based on the decreased cost of decoding nucleotide sequences, serving as the basis for discovering genes with specific functions in various genetic resources.
Transposon prediction using repeat models and classification according to structural characteristics, extraction of Simple Sequence Repeats (SSRs or Microsatellites) on the genome sequence, and primer development are performed. Gene function prediction based on homology search is possible for the final gene set. Hybrid assembly is a method of assembling by combining different sequencing technologies, which is superior in terms of completeness and accuracy. Long high-fidelity (HiFi) reads of our own equipment, PacBio Sequel II, are produced with an average length of 13.5 kb and 99.9% accuracy, and a highly accurate draft genome is provided by assembling with Illumina short reads. After determining the gene model through reference to a closely related species or RNA-seq or Iso-Seq, the assembled draft genome predicts biological function and protein domain information through homology search with UniProt, NCBI nr, InterProScan, and Pfam databases.
Application
- Phylogenetic classification
- Species diversity studies
Sample/Laboratory Information
| Sample Requirement | gDNA > 2µg (min 500 ng), 20 ng/µl, DIN > 6 |
|---|---|
| Library Kit | Illumina, PacBio |
| Sequencing Platform | Illumina NovaSeq6000, PacBio Sequel Ⅱ, PacBio Revio |
| Recommended Sequencing Depth | ≥ 10 milion read pair per sample |
Bioinformatics
- Workflow
-
-
Standard Analysis
Genome denovo assembly
Genome annotation
Error correction
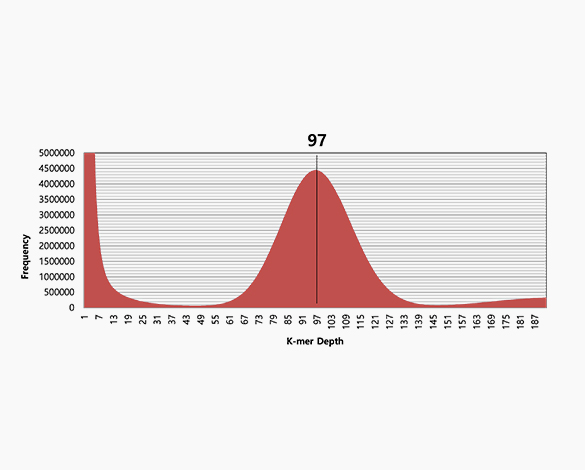
k-mer analysis
-
Advanced Analysis
Construction of mitochondrial genome
Construction of chloroplast genome
Phylogenetic analysis
No content
Reference
[1] Yim, H. S. et al., (2014). Minke whale genome and aquatic adaptation in cetaceans. Nature genetics, 46(1), 88–92.
[2] Cho, Y. S. et al., (2013). The tiger genome and comparative analysis with lion and snow leopard genomes. Nature communications, 4, 2433.
[3] Choo, J. H. et al., (2016). Whole-genome De novo sequencing, combined with RNA-Seq analysis, reveals unique genome and physiological features of the amylolytic yeast Saccharomycopsis fibuligera and its interspecies hybrid. Biotechnology for biofuels, 9, 246.
[4] Kim, J. et al., (2020). Whole-genome, transcriptome, and methylome analyses provide insights into the evolution of platycoside biosynthesis in Platycodon grandiflorus, a medicinal plant. Horticulture research, 7, 112.
[5] Park, S. G. et al., (2021). Draft Genome Assembly and Transcriptome Dataset for European Turnip (Brassica rapa L. ssp. rapifera), ECD4 Carrying Clubroot Resistance. Frontiers in genetics, 12, 651298.
Whole Exome Sequencing
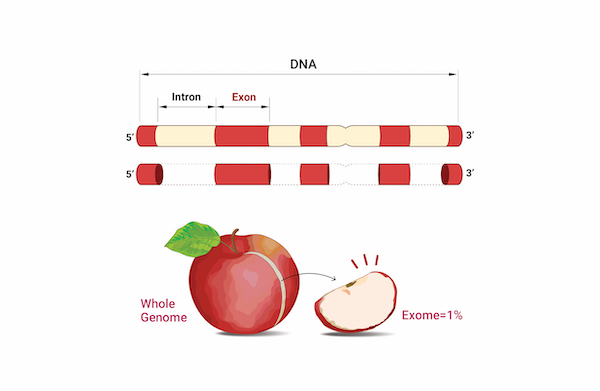
As the human genome information becomes more extensive and genetic information about various diseases and phenomena is established, it has become possible to acquire more information through whole exome sequencing. High-depth Germline and Somatic mutation information is provided for the Exome region (less than 2% of the entire genome in humans), which directly codes for proteins in the entire genome. The annotated variants are categorized and classified by function, providing clinically significant mutation information.
Application
- Genetic disease-related studies
- Cancer research and drug development
- Pathogenic mechanism and molecular characterization
Sample/Laboratory Information
| Sample Requirement | gDNA 500ng (minimum 50ng) 10 ng/µl, DIN > 6 |
|---|---|
| Library Kit | SureSelect(Agilent), Twist, etc |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | ≥100X (For tumor, ≥200X) |
Bioinformatics
- Workflow
-
-
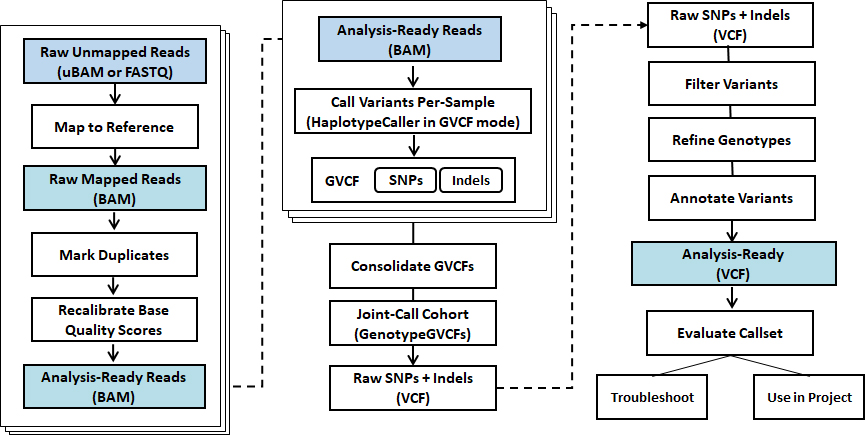
Standard Analysis
Germlin & Somatic
Read mapping + BQSR, VQSR
Variant calling (SNV, Indels)
Variant annotation
-
Advanced Analysis
Oncoplot
No content
No content
No content
No content
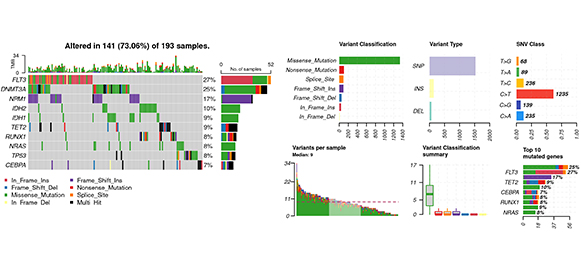
Features & Benefits
- Variant detection applicable in various fields
- High coverage of sequence information in protein regions
- Faster and easier data production compared to whole genome analysis techniques
Considerations
- Amount of coverage to be secured for the produced data
- Variability may exist depending on the hybrid capture kit
- Areas where coverage cannot be secured (Repeat elements, Tandem repeats and Pseudogenes)
- The existence of regions with extreme GC ratios
Reference
[1] Phi, J. H. et al., (2018). Genomic analysis reveals secondary glioblastoma after radiotherapy in a subset of recurrent medulloblastomas. Acta neuropathologica, 135(6), 939–953.
[2] Chang, Y. H. et al., (2016). Use of whole-exome sequencing to determine the genetic basis of signs of skin youthfulness in Korean women. Journal of the European Academy of Dermatology and Venereology, 31(3), e138–e141. https://doi.org/10.1111/jdv.13904
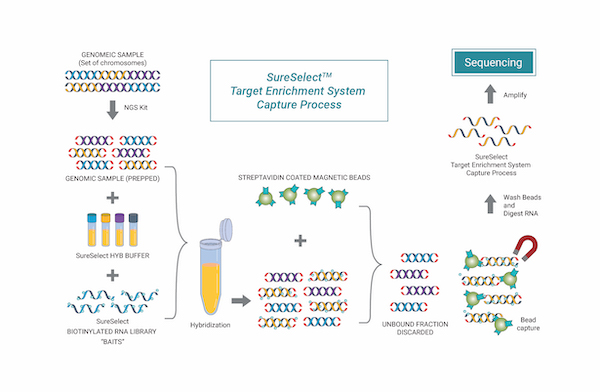
Targeted Sequencing
Targeted sequencing has the advantage of allowing for high-resolution mutation analysis, making it possible to detect somatic mutations in tissue samples with low purity, such as FFPE (Formalin Fixed Paraffin Embedded) samples. It is the most appropriate method for clinical laboratories and is commonly used in disease-related gene discovery, where the target genes and regions of interest are clearly defined.
* Liquid biopsy is a service that accurately analyzes mutations in cancer cell-free DNA floating in the blood with only a small amount of blood sample. It can detect cancer somatic mutations with reproducibility even at a detection limit of less than 0.01%. Each analysis performed from a single blood tube is used to analyze single nucleotide variants and short indels that frequently occur in the sample.
Application
- Tumor mutation burden(TMB)
- Disease or phenotype-related studies
- Detection of low-frequency alleles and rare variants
- Identification of causative novel or inherited mutations
- Drug response and discovery of therapeutic targets
Sample/Laboratory Information
| Sample Requirement | gDNA > 700ng (minimum 500 ng), 10 ng/µl, DIN > 6 |
|---|---|
| Library Kit | Agilent, Twist |
| Sequencing Platform | Illumina NovaSeq6000 |
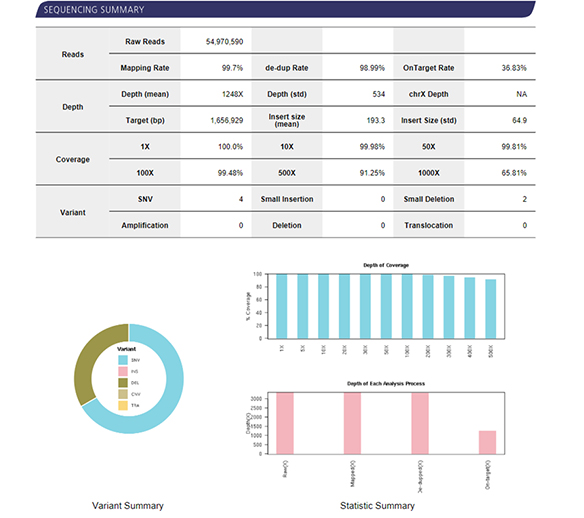
| Recommended Sequencing Depth | 200X, 500X, 1000X |
Bioinformatics
- Workflow
-
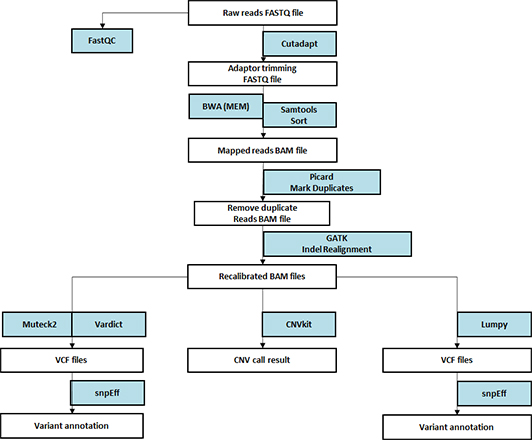
-
Standard Analysis
Read mapping
Variant calling
Variant annotation
-
Advanced Analysis
Genomic rearrangement
Tumor mutation burden (TMB), Microsatellite instability (MSI)
Features & Benefits
- Variant detection for Targeted Sequencing Panels for clinical testing purposes
- Variant detection confirmed through national cancer information projects
- Providing guidance in the decision-making process for disease treatment of patients by clinicians through variant discovery.
Reference
[1] Kim, H. S. et al., (2018). Association of PD-L1 Expression with Tumor-Infiltrating Immune Cells and Mutation Burden in High-Grade Neuroendocrine Carcinoma of the Lung. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer, 13(5), 636–648.
- Theragen Bio
- Business Number : 816-86-01640
- CEO : Jin Yeob, Go / Soon Myung, Paik
- B-4F, 145, Gwanggyo-ro, Yeongtong-gu, Suwon-si, Gyeonggi-do, Republic of Korea
- Tel : +82+1522-2382
- Fax : +82-31-288-1293
COPYRIGHT(C) THERAGEN BIO CO.,LTD. ALL RIGHTS RESERVED.
