Services
서비스
NGS
Targeted Methylation Sequencing
DNA methylation이 가능한 4개의 뉴클레오타이드 중 메틸화의 레벨을 CpG island, Promoter region 등의 게놈 영역에 대해 확인할 수 있으며,
Whole genome bisulfite sequencing 데이터보다 작은 양의 시퀀싱 데이터만으로 주요한 영역의 Methylation pattern을 확인할 수 있습니다.
DNA methylation 데이터는 유전자 발현, 배아 발달, 세포 증식, 분화 및 염색체 안정성 등의 생물학적 현상 연구에 중추적인 역할을 합니다.
비정상적인 DNA methylation은 암과 같은 인간 질병으로 이어지는 DNA 항상성 상실 및 게놈 불안정성과도 관련이 있습니다.
적용 가능 연구
- Profiling dysregulation of DNA methylation
- Studying genotype-environment interactions and disease-related patterns
- Investigation of individual variation in disease susceptibility
샘플&실험정보
| Sample Requirement | gDNA > 3µg (minimum 2µg),60ng/µl, DIN > 6 |
|---|---|
| Library Kit | Agilent |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | ≥ 20 million read pairs per sample |
생명정보학 분석
- Workflow
-
Standard Analysis
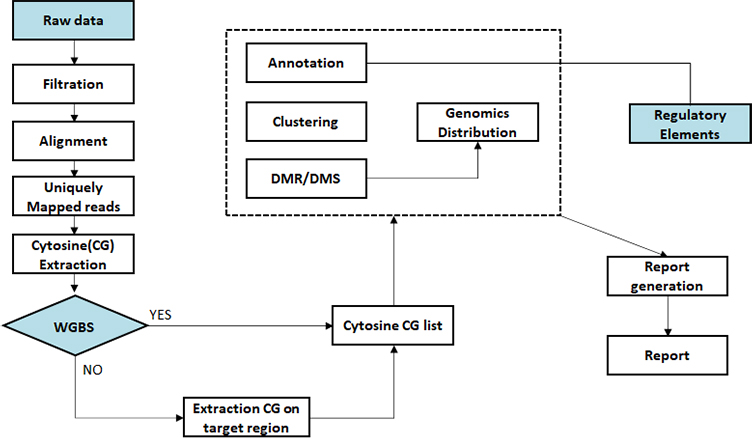
Sequencing data preprocessing
Alignment bisulfite sequencing data to genome
Methylation calling
Differential methylated region (DMR) analysis
Differential methylated region (DMR) annotation
Reference
[1] Shin, H. Y. et al., (2020). Alteration in global DNA methylation status following preconditioning injury influences axon growth competence of the sensory neurons. Experimental neurology, 326, 113177.
[2] Kato, N. et al., (2015). Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nature Genetics, 47(11), 1282–1293. https://doi.org/10.1038/ng.3405
Bisulfite Sequencing
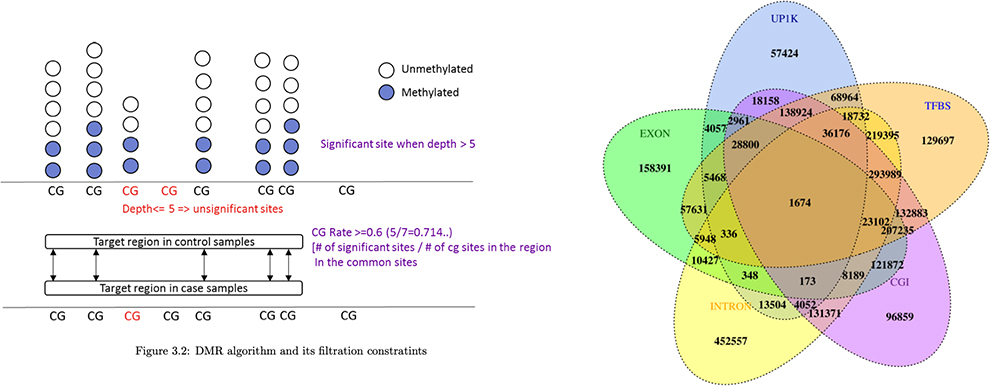
Bisulfite sequencing 을 통해 전장 유전체 상에 존재하는 Cytosine context (CpG, CHG, CHH) 에 대한 DNA methylation level 을 확인할 수 있습니다.
후성 유전적 현상을 Molecular 수준에서 연구하는 데 사용되며, 샘플 및 그룹 간 Differential methylated region을 확인하고
해당 영역의 Genomic contents를 Annotation하여 유전자의 발현에 미치는 영향을 확인할 수 있습니다.
테라젠바이오는 도라지에서 Whole genome bisulfite sequencing 데이터를 이용해 Platycosides biosynthesis를 연구한 바 있습니다.
적용 가능 연구
- Profiling DNA methylation
- Studies on phenotypic diversity and evolution in plants and animals
- Understanding of epigenetic alternations of pathogenetic pathways
- Early diagnosis of diseases and cancers through the detection of DNA hypermethylation or hypomethylation
샘플&실험정보
| Sample Requirement | gDNA > 3µg (minimum 2µg),60ng/µl, DIN > 6 |
|---|---|
| Library Kit | Swift |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | For effective sequencing depth 50X |
생명정보학 분석
- Workflow
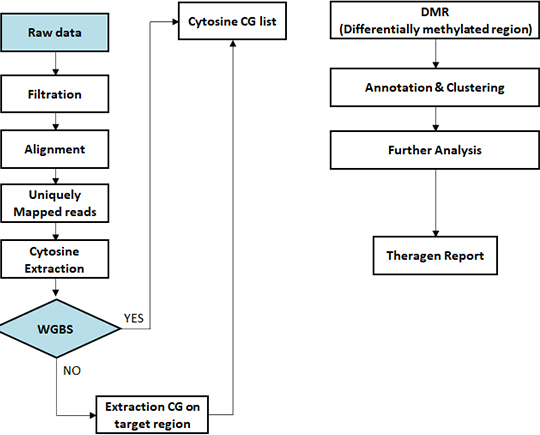
-
Standard Analysis
Reference building for non-human species
Sequencing data preprocessing
Alignment bisulfite sequencing data to genome
Methylation calling
All cytosine context (CpG, CHG, CHH)
Differential methylated region (DMR) analysis
Differential methylated region (DMR) annotation
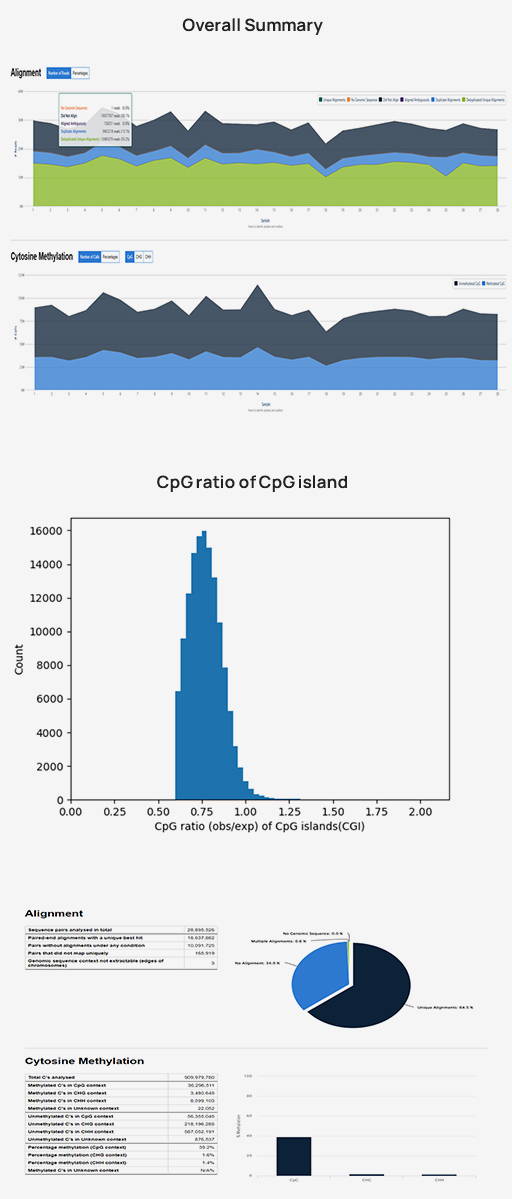
-
Advanced Analysis
Methylation rate (CpG, CHG, CHH)
Methylation pattern (Upstream, Genebody, Downstream)
Density of differentially methylated cytosines (DMCs)
No content
No content
No content
No content
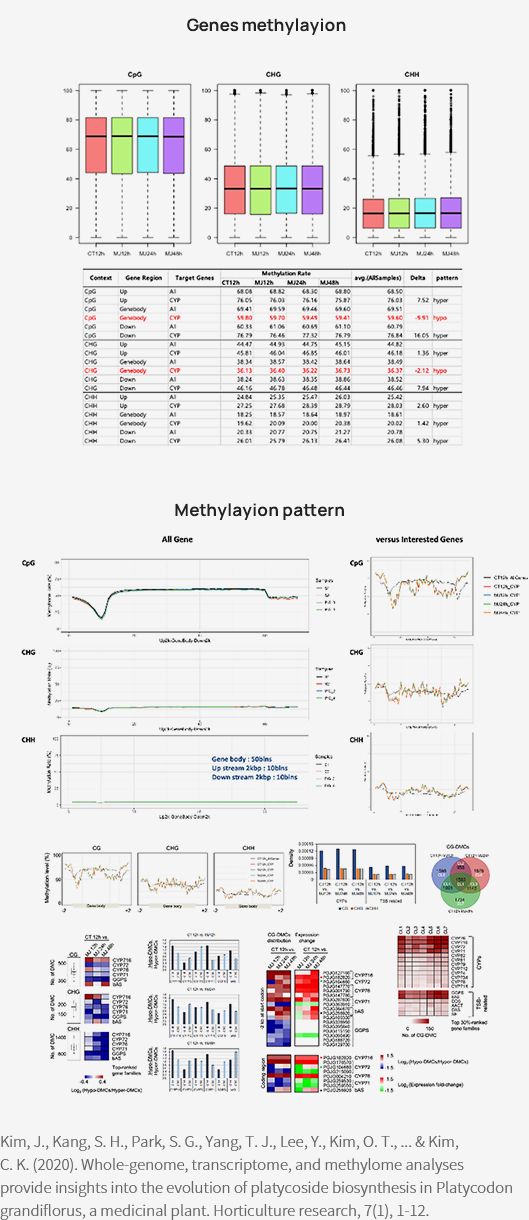
Reference
[1] Kim, J. et al., (2020). Whole-genome, transcriptome, and methylome analyses provide insights into the evolution of platycoside biosynthesis in Platycodon grandiflorus, a medicinal plant. Horticulture research, 7, 112.
ChIP Sequencing
ChIP sequencing은 염색질 면역 침전(ChIP)과 NGS을 결합한 시퀀싱 방법이며, Bioinforamtics 분석을 통해 DNA 관련 단백질의 결합 부위를 식별할 수 있습니다.
식별된 결합 부위는 각 세포 유형의 후성 유전적 상태를 정확하게 예측할 수 있으며, 세포 과정의 조절, 세포의 분화 과정 또는 질병 진행 메커니즘을 이해하는 데 필수적입니다.
ChIP sequencing은 면역침전법을 사용함에 따라 필연적으로 기술적 편향이 발생할 수 있기 때문에,
IgG와 같은 배경 제어 및 전산 분석 방법을 사용한 정밀한 실험 설계를 통해 줄일 수 있습니다.
샘플&실험정보
| Sample Requirement | gDNA > 3µg (minimum 2µg) |
|---|---|
| Library Kit | Illumina |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | ≥ 20 million reads per sample |
생명정보학 분석
- Workflow
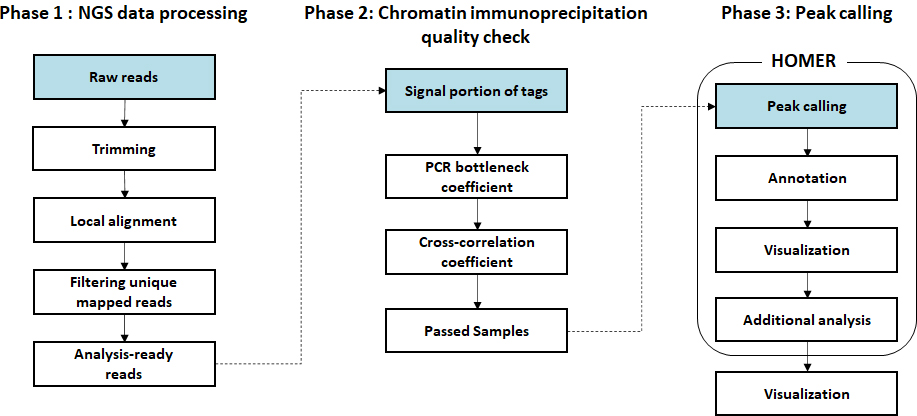
-
Standard Analysis
Sequencing data preprocessing
Local alignment to genome
Chromatin immunoprecipitation quality check
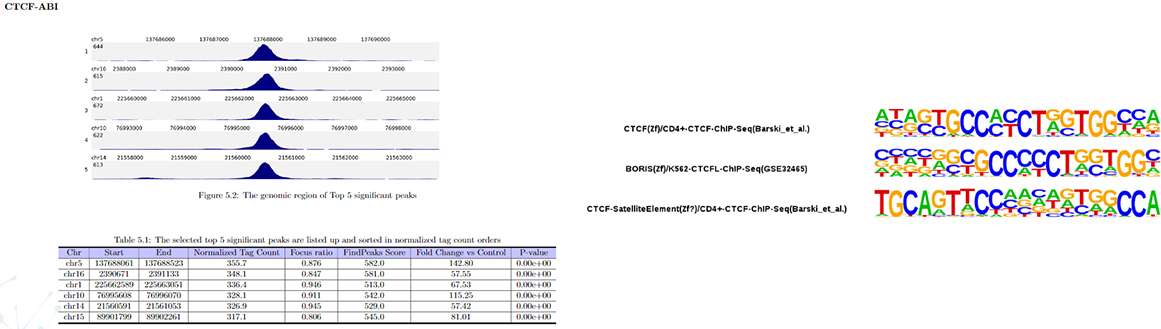
Peak calling
Peak annotation
Transcription factor motif analysis