

Services
서비스
NGS
Total/mRNA Sequencing
테라젠바이오는 인체, 오가노이드, 동식물, 제노크래프트 등 다양한 샘플들로부터 정상군과 대조군의 차별적 유전자 발현량 분석 (Differentially Expressed Genes Analysis) 을 제공하며,
이를 기반으로 유전자 기능 연구에 필요한 단초를 제공해 드립니다. 생물학적인 특성에 관련된 유전자들이 어떠한 조건 (예: 정상세포 vs 암세포)에서 통계적 의미를 가지는지
Gene set enrichment analysis (GSEA) 분석이 가능합니다. 기존에 동정되지 않았던 전사체나 대체접합 정보 (Alternative splicing) 및 융합 유전자 정보 (Gene fusion) 등도 제공합니다.
샘플&실험정보
| Sample Requirement | 0.1 ~ 1µg Total RNA 0.1 ~ 4µg Total RNA, RIN ≥ 8 |
|---|---|
| Library Kit | Illumina, TaKaRa, Qiagen |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | 20M (million reads) |
생명정보학 분석
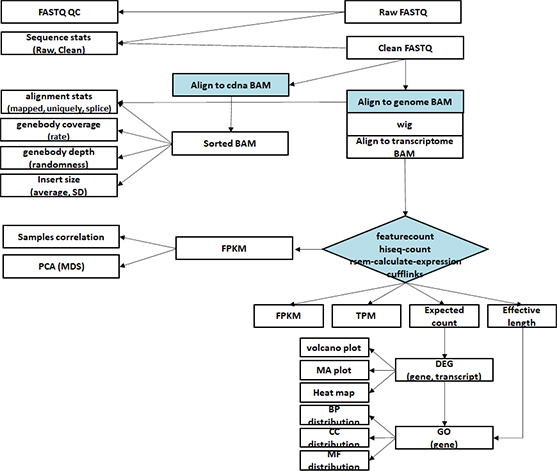
- Workflow
-
-
Standard Analysis
Functional annotation
Alternative splicing analysis
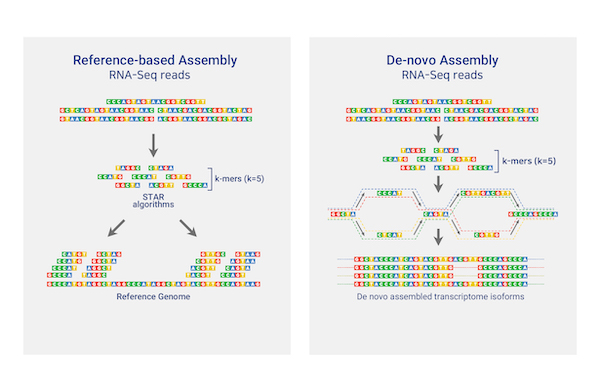
RNAseq De novo assembly
Differential expression genes (DEGs)
GO analysis
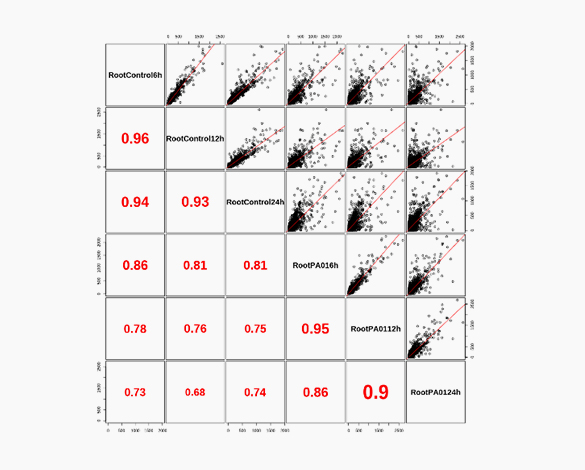
Heatmap
-
Advanced Analysis
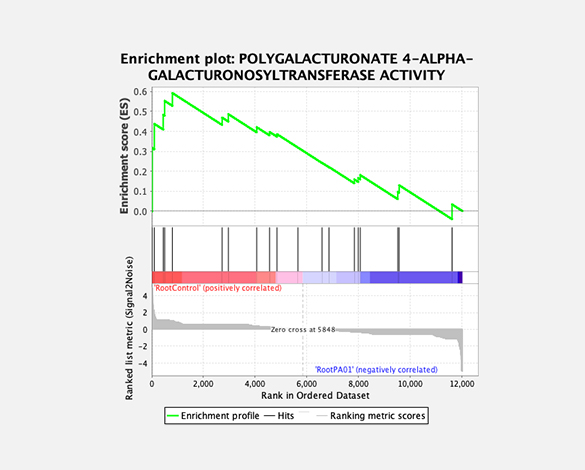
Gene set enrichment analysis (GSEA)
Gene fusion analysis
No content
No content
No content
Features & Benefits
- 생물학적 변화를 모두 포착할 수 있는 유전자 레벨의 광범위한 데이터를 획득 가능
- 유전자 레벨에서 알려지거나 알려지지 않은 정보들을 획득 가능
- 참조 유전체 정보가 없어도 유전자 레벨의 정량적 측정이 가능
Reference
[1] Nakayama, M. et al., (2020). Loss of wild-type p53 promotes mutant p53-driven metastasis through acquisition of survival and tumor-initiating properties. Nature communications, 11(1), 2333.
[2] Hong, E. et al., (2020). Inhibition of TGF-β signalling in combination with nal-IRI plus 5-Fluorouracil/Leucovorin suppresses invasion and prolongs survival in pancreatic tumour mouse models. Scientific reports, 10(1), 2935.
[3] Cha, S. et al., (2021). Differential activation mechanisms of two isoforms of Gcr1 transcription factor generated from spliced and un-spliced transcripts in Saccharomyces cerevisiae. Nucleic acids research, 49(2), 745–759.
[4] Yun, S. M. et al., (2013). PPP1R1B-STARD3 chimeric fusion transcript in human gastric cancer promotes tumorigenesis through activation of PI3K/AKT signaling. Oncogene, 33(46), 5341–5347. https://doi.org/10.1038/onc.2013.472
[5] Yoon, K. et al., (2013). Comprehensive genome- and transcriptome-wide analyses of mutations associated with microsatellite instability in Korean gastric cancers. Genome Research, 23(7), 1109–1117. https://doi.org/10.1101/gr.145706.112
[6] Kim, K. et al., (2018). Pathway profiles based on gene-set enrichment analysis in the honey bee Apis mellifera under brood rearing-suppressed conditions. Genomics, 110(1), 43–49. https://doi.org/10.1016/j.ygeno.2017.08.004
[7] Sakai, E. et al., (2017). Combined Mutation of Apc, Kras, and Tgfbr2 Effectively Drives Metastasis of Intestinal Cancer. Cancer Research, 78(5), 1334–1346. https://doi.org/10.1158/0008-5472.can-17-3303
[8] Park, Y. et al., (2020). Destablilization of TRAF6 by DRAK1 Suppresses Tumor Growth and Metastasis in Cervical Cancer Cells. Cancer Research, 80(12), 2537–2549. https://doi.org/10.1158/0008-5472.can-19-3428
[9] Lee, J. et al., (2021). Brassinosteroid‐BZR1/2‐WAT1 module determines the high level of auxin signalling in vascular cambium during wood formation. New Phytologist, 230(4), 1503–1516. https://doi.org/10.1111/nph.17265
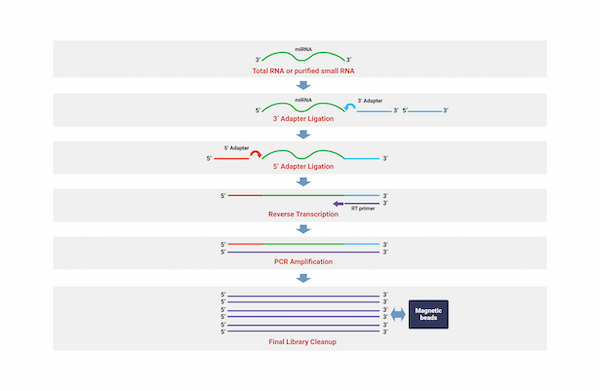
Small RNA Sequencing
세포성장, 조직 분화, 세포 자살, 바이러스 감염과 같은 다양한 생물학적 과정을 조절하는 비발현 작은 RNA(small non-coding RNA, ncRNA)를 동정하는 기술을 통해
수천개의 micro RNA(miRNA)와 같은 Small ncRNA서열에 대하여 광범위한 수준으로 분석해 낼 수 있습니다.
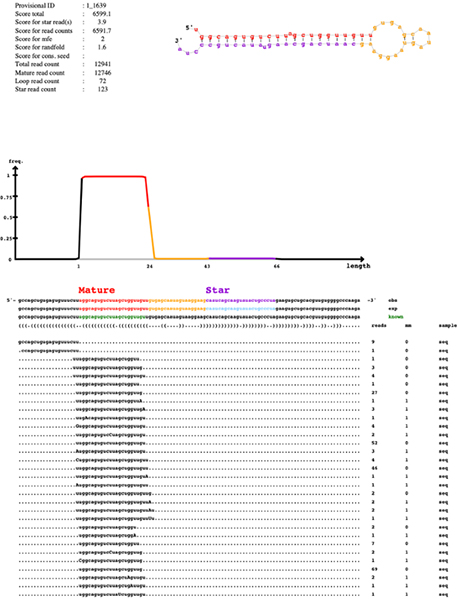
아직 알려지지 않은 miRNA 뿐만 아니라 샘플들 내에서 차별적으로 발현되는 작은 RNA 단위들을 동정함으로써 연구자들에게 필요한 정보를 제공합니다.
miRNA seed 영역 변이를 탐색하며, 해당 종의 유전자 정보를 활용하여 발현량을 측정합니다.
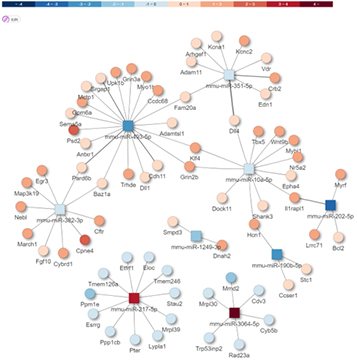
또한, 다양한 Database 정보를 통합하여 miRNA가 Target 하는 유전자 정보 제공하며, 각각의 miRNA가 Target 하는 유전자에 대한 정보를 제공합니다.
샘플&실험정보
| Sample Requirement | Total RNA > 1µg (minimum 100 µg), 20 ng/µl, RIN > 6 |
|---|---|
| Library Kit | PerkinElmer |
| Sequencing Platform | Illumina NovaSeq6000 |
| Recommended Sequencing Depth | ≥ 20 million read counts per sample |
생명정보학 분석
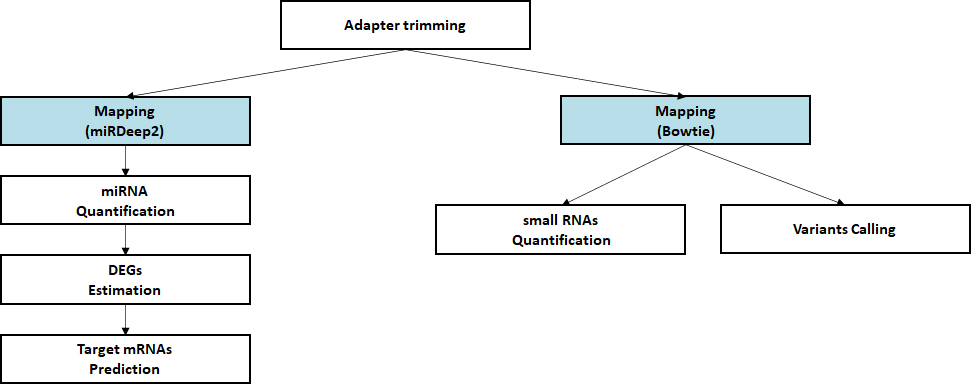
- Workflow
-
Standard Analysis
Alignment against reference genome
Quantification of small RNA
Discovery of novel/known miRNAs
Estimation of differential expressed miRNAs
Prediction of target mRNA
-
Advanced Analysis
miRNA-mRNA Integrated analysis
No content
No content
No content
No content
No content
Reference
[1] Lee, J. Y., Ryu, D. S., Kim, W. J., & Kim, S. J. (2016). Aberrantly expressed microRNAs in the context of bladder tumorigenesis. Investigative and clinical urology, 57 Suppl 1(Suppl 1), S52–S59.
[2] Kim, M. C. et al., (2014). Identification and characterization of microRNAs in normal equine tissues by Next Generation Sequencing. PloS one, 9(4), e93662.
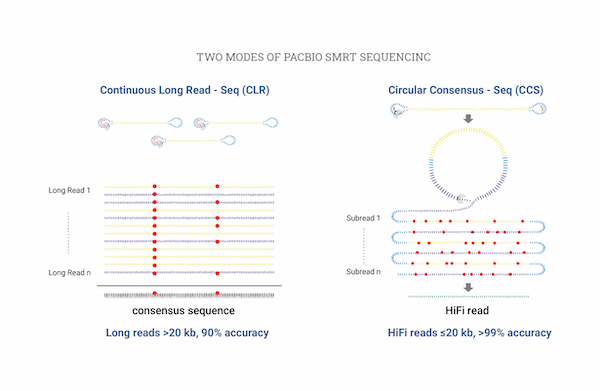
Isoform Sequencing (de novo)
Isoform sequencing은 Short-read에서 발생하는 오류를 방지하여 Full length isoform을 시퀀싱 및 분석을 할 수 있습니다.
PacBio 기반 Long-read 서열을 이용하여 Clustering 및 Annotation을 하여 Unigene을 만들수 있습니다.
또한, illumina 기반 Short-read 서열을 이용하여 Unigene에 대한 RNA-seq Expression을 계산할 수 있습니다.
적용 가능 연구
- Alternative splicing
- Alternative polyadenylation (APA)
- Fusion transcripts
- Long non-coding RNAs (lncRNAs)
- Isoform phasing
- Genome annotation
샘플&실험정보
| Sample Requirement | RNA > 3µg (min 1µg),100ng/µl, RIN > 8 |
|---|---|
| Library Kit | SMRT |
| Sequencing Platform | PacBio Sequel Ⅱ, PacBio Revio |
| Recommended Sequencing Depth | ≥ 15Gb per sample ≥ 100 million read pairs per sample *Depth may vary depending on species |
생명정보학 분석
- Workflow
-
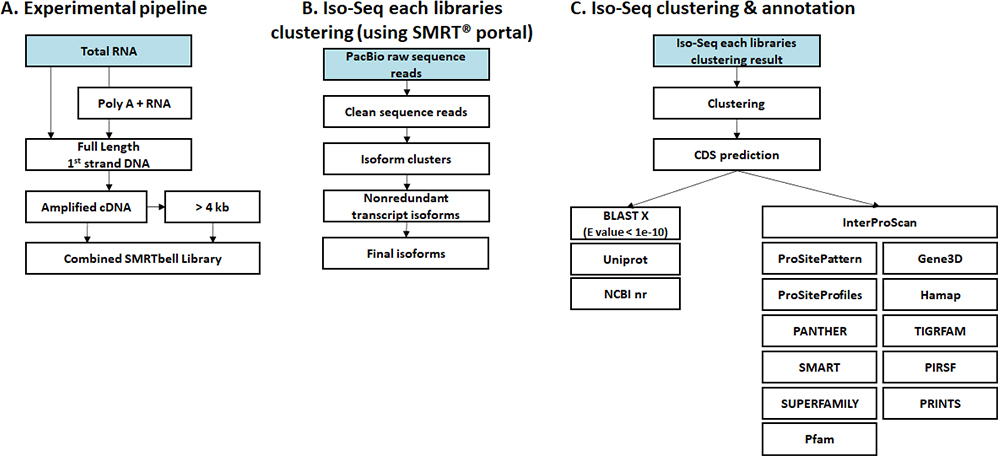
-
Standard Analysis
Clustering & Annotation
Read mapping
Expression
Differential expression genes (DEGs)
Reference
[1] Jo, I. H. et al., (2017). Isoform Sequencing Provides a More Comprehensive View of the Panax ginseng Transcriptome. Genes, 8(9), 228.
[2] Hong, C. P.et al., (2022). Long-read transcriptome sequencing provides insight into lignan biosynthesis during fruit development in Schisandra chinensis. BMC genomics, 23(1), 17.
